【美华社 2025年12月3日 马里兰州 讯】美国食品药品监督管理局(FDA)于当地时间12月3日举办了关于《生物类似药用户费用法案》(BsUFA)重新授权的公开会议,启动了 2028–2032 财年 BsUFA 授权流程的公众意见征集和行业咨询阶段。此次行动标志着FDA在推动高质量生物药审批、优化研发路径以及释放市场潜力方面迈出了关键一步。
百亿资金助力审批与创新

美国食品药品监督管理局(FDA)药品评审与研究中心 副中心主任 Mike Davis 在开场中指出,BsUFA自2012年首次实施以来,已成为扩大生物类似药可及性、提升审批效率和保障药品安全的重要工具。自2015年以来,生物类似药累计为美国医疗体系节省约560亿美元,其中2024年单年节省约200亿美元。通过 BsUFA,FDA已批准78个生物类似药,建立了稳定、可预测的审批流程,为患者提供更多可负担的生物药选择。

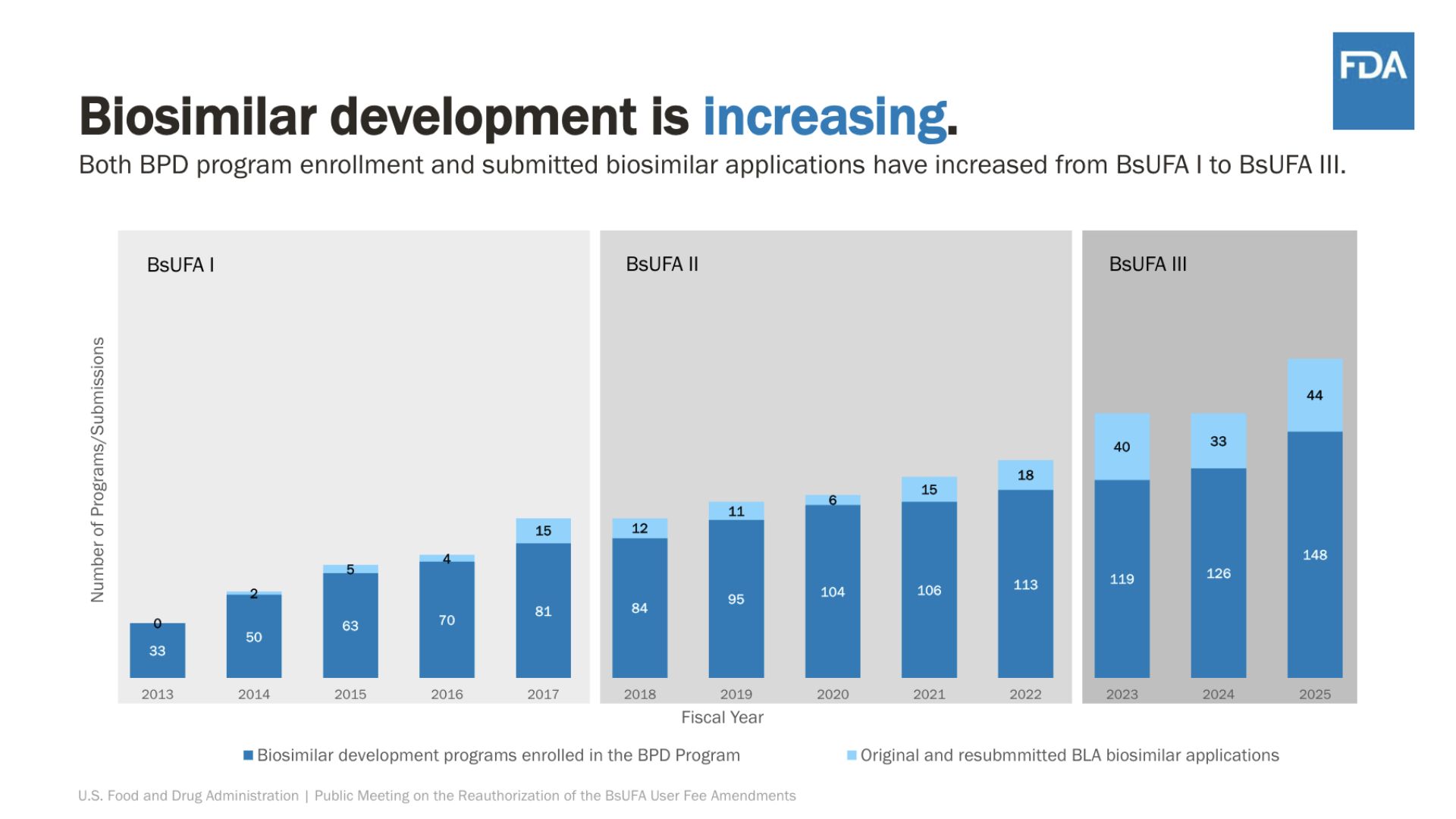
美国药管局 药品评审与研究中心 战略分析办公室主任Andrew Kish介绍了 BsUFA 的历史与三阶段发展:
- BsUFA I:建立用户费用结构和初步审批流程,参考PDUFA(处方药用户费用法案)模式。
- BsUFA II:优化费用结构、引入新的评审承诺,提高审批效率。
- BsUFA III:新增补充申请类别、风险分析及制造相关审查时间表,启动监管科学试点项目,提升可互换药物开发指导。
会议显示,用户费用收入占 BsUFA 项目总预算约61%,主要用于支持审批人员和流程管理。BsUFA III 的绩效表现稳定,审批流程、会议管理和数据发布均有提升。
行业关注审批效率与市场准入
行业代表在会议中分享了对 BsUFA IV 的期待与建议。

生物类似药论坛(The Biosimilars Forum)执行董事 Juliana Reed 和 生物类似药委员会 科学与监管事务高级总监 Scott Kuzner 指出,生物类似药不仅降低药品成本,还为美国医疗体系节约巨额开支。行业关注点包括:
- 审批时间仍偏长,尤其在 FDA 不再普遍要求临床等效性研究后。
- 需要提升沟通效率、提高透明度、加快检查流程。
- 支持 FDA 以分析数据为主的监管趋势,减少不必要的临床试验。
- 探讨全球统一参比药(Single Global Comparator)和优化药物-器械组合产品的人因评估。
- 强调资金稳定性,避免触发用户费用释放条件(spending trigger)导致资源中断。
美国创新药物制造商协会(PhRMA)科学与监管事务高级总监 Sean Hilscher 则代表创新药企强调,BsUFA IV应保持科学为基础的监管体系、稳定的沟通机制、透明的审评时间表及可量化的KPI,以支持 FDA 的长期资源保障和审批可预测性。
Teva Pharmaceuticals代表Aaron Josephson指出,生物类似药不仅降低成本,还提升患者生产力,对社会经济有积极影响。他建议改进审批流程中的信息请求时机、减少“late-cycle labeling comments”、提前安排检查,并确保监管科学项目资金使用透明,同时将最终批准签署权交由最了解产品的审批办公室(OTBB),以提高效率。
公众参与与流程透明
此次会议也是 BsUFA 重新授权流程的第一步。公众可通过提交书面意见参与,截止日期为2026年1月2日。技术谈判预计将于2026年春季开始,并在夏季完成,随后进入 FDA 内部审批及向国会提交新授权草案的阶段(预计2027年)。会议期间,FDA承诺每月至少一次与利益相关方进行咨询,并公开谈判纪要,确保透明和可追踪。
商业与创新机遇
对于生物医药企业和创业者而言,BsUFA IV的重新授权不仅提供资金与资源支持,还意味着加速审批、优化研发路径和开拓市场的战略窗口期。监管科学试点项目为企业提供早期指导,降低审批不确定性;高效审批和可互换药物获批将进一步拓展市场空间,为创新企业和投资人带来潜在收益。
此次公开会议为行业提供了深入了解 FDA 审批机制和未来政策方向的机会,也为生物类似药市场的下一步发展定下了基调。BsUFA IV重新授权流程的启动,标志着美国生物医药产业在研发效率、市场竞争力与患者可及性方面迎来新的发展机遇。

会议由FDA药品评价与研究中心的Jonathan Collins主持。